EXtra-xwiz¶
EXtra-xwiz is a command-line tool for automated processing of the calibrated serial crystalography data. It is a semi-automatic pipeline wrapped around CrystFEL and it utilises SLURM for parallel computing.
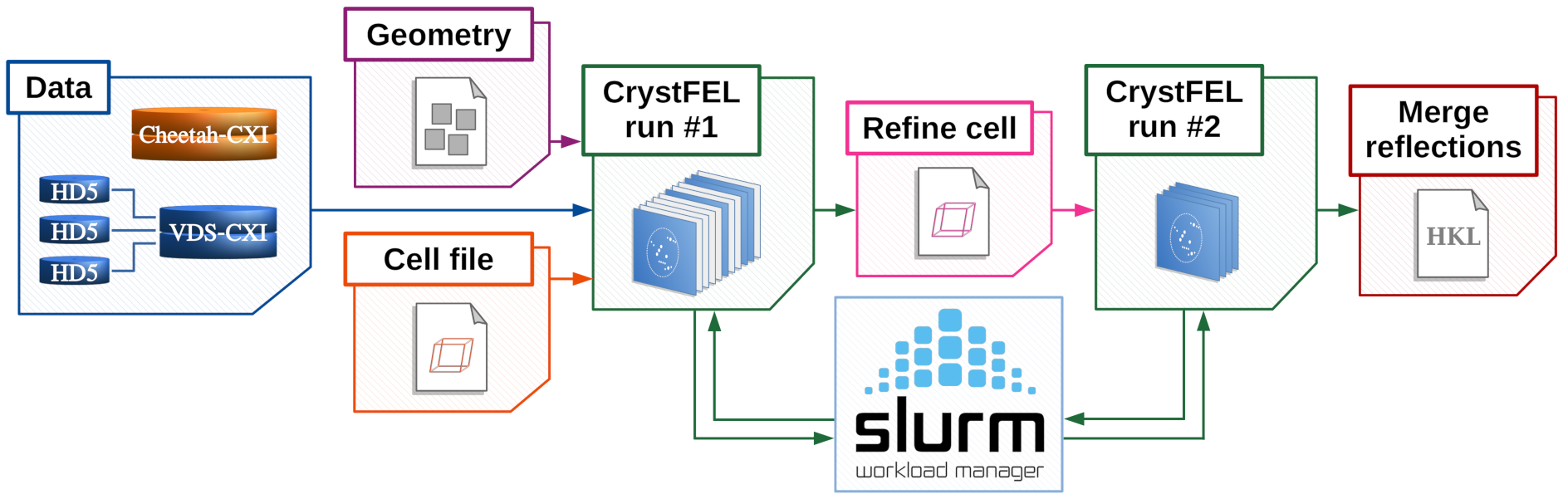
The basic functionality of the tool is explained here.
Installation¶
EXtra-xwiz is pre-installed in the EuXFEL software folder on Maxwell, currently in a virtual environment. To activate it, simply type
module load exfel EXtra-xwiz/beta
Getting and setting a configuration¶
The workflow configuration determines how the steps of a typical SFX workflow shall be run. The first call of the tool::
xwiz-workflow
Will do nothing except writing a template configuration file to the current folder.
Luckily, there are only a few configuration items that have to be adapted by the user, because no reasonable default exists. Open the freshly created file xwiz_conf.toml with a text editor and change the right-hand parts of the definitions as required, e. g. proposal number, run and so on.
If configuration file with more parameters is desired -adv / --advance-config option can be used:
xwiz-workflow --advance-config
Configuration File¶
Generally the configuration is structured into groups, addressing different categories or aspects of the pipeline usage, such as input file retrieval, CrystFEL program parameters or SLURM (distributed computation) set-up.
A few specific aspects of the configuration values may not be obvious, let's look at the "data", "crystfel", "geom, and "slurm" groups.
[data]¶
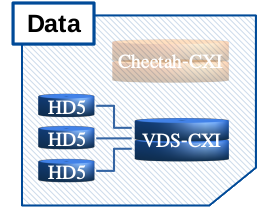
[data]
proposal = 900145
runs = 752
n_frames_total = 10000
vds_names = ["jf_752_vds.cxi"]
list_prefix = "p900145_r0752"
proposal¶
Proposal number which identifies your beam-time. Accessing calibrated/corrected detector data, Xwiz will expand this to the path of the proposal "proc" folder containing the runs you will work with.
runs¶
Single data collection run or a list of run numbers that shall be processed within one Xwiz session. To add another run to the above example, change to e. g.: [752,753]
vds_names¶
List of one or more virtual data set (VDS) file names. Any valid/existing path - absolute or relative - may be used. If the string(s) are only names, the path is interpreted as the current working directory (i. e. the folder from where Xwiz was started).
There are two ways to work with VDS files:
- Create them from the original data in runs, typically at the first processing session.
- the runs parameter is essential
- vds_names point to target file names of VDS files to be created
- there must be one name for each run
- e.g. in case of two runs:
["jf_752_vds.cxi", "jf_753_vds.cxi"]
- Use existing VDS files.
- The software first looks if the given files are already present
- if so, the runs setting is ignored and the VDS will be re-used
n_frames_total¶
The total number of frames to be processed, over all runs. Having n_frames_total < actual sum of frames is a way to truncate processing. The case n_frames_total > actual sum of frames will take the actual sum.
list_prefix¶
All output files of Xwiz will start with this tag, it thus identifies the session.

[crystfel]
# Available versions: '0.8.0', '0.9.1', '0.10.0', 'cfel_dev'
version = 'cfel_dev'
version¶
Version number of the CrystFEL release to use. The need to switch to older releases may be given by the reproduction or comparison purposes. cfel_dev points to the latest builds, prior to the upcoming release.

[geom]
file_path = "jungfrau8_p900145_v1_vds.geom"
file_path¶
Relative or absolute path to an existing CrystFEL-format geometry file appropriate for the detector geometry at the time of data collection.
Geometry files can come in two fashions, with either Cheetah-compatible or VDS-compatible pixel data references. The main difference concerns fused vs. stacked pixel data from multi-module detectors (AGIPD, JF4M). As per Cheetah layout, all pixels of an image-frame are in one set with just the dimensions (ss, fs), whereas in VDS layout there is an additional dimension containing the module index.
If you have the correct geometry in terms of metrics, but the file refers to the wrong pixel layout, Xwiz will auto-correct these layout references to match the used data file. For instance, a geometry file stemming from usage with Cheetah will be auto-converted to VDS layout if Xwiz is run with a VDS file (default).
[slurm]¶

[slurm]
# Available partitions: 'all', 'upex', 'exfel'
partition = "all"
duration_all = "2:00:00"
n_nodes_all = 10
duration_hits = "0:30:00"
n_nodes_hits = 4
partition¶
Points to the corresponding Maxwell partition of HPC nodes to be used. Default all would suite most needs.
External users could also choose upex - or in case of dedicated beam-time reservations something like upex_PPPPPP (with PPPPPP as the proposal number, padded to 6 digits with 0's, e.g. for proposal 2222 this would be upex_002222).
duration_all¶
Warning
In practice, processing that takes longer to finish than the allocated time will be aborted!
(resp. duration_hits) The format reads "H:MM:SS" and defines for how long the HPC nodes shall be allocated.
n_nodes_all¶
(resp. n_nodes_hits) Specifies how many HPC nodes to employ. Hard limits are set by the partition resp. reservation.
Info
The _all vs. _hits distinction allows to have separate values for the processing of all frames in the first phase versus the re-processing of a subset of crystal "hit" frames (indexable due to Bragg diffraction patterns contained) in the second phase of processing.
Running the Fully Automatic Mode¶
Provided that a good starting estimate for the crystal unit cell is available, one can create a CrystFEL unit cell file in the current folder, like this::
CrystFEL unit cell file version 1.0
lattice_type = tetragonal
centering = P
unique_axis = c
a = 79.1 A
b = 79.1 A
c = 37.9 A
al = 90.00 deg
be = 90.00 deg
ga = 90.00 deg
and specify the saved file name in the configuration.
To process the diffraction data, one can then enter
xwiz-workflow -a
and let everything run based on the configuration, ending up with the merged structure factor intensities and their crystallographic FOM vs. resolution- shell tables.
Running the Interactive Mode¶
Essentially, interactive mode means that all items of the configuration file (including advanced configuration parameters) are displayed for confirmation at a prompt at run-time. This is meant to allow for quick re-confirmation or modification of the workflow parameters, without the need of changing the configuration persistently by file editing.
The interactive mode is actually the default, meaning it is chosen when the tool is called without command-line argument. Hence, type:
xwiz-workflow
and you will enter the interactive mode.
Output Files¶
The following files can be found in the current folder, from where the tool was run, after the workflow has finished:
- A virtual data set for
indexamajig(only in case that a non-existing file was referred to in the config) - Stream files from
indexamajig - A file
<prefix>_hits.lstcontaining the numbers of detector frames that could be indexed (a sub-set of all frames) - The unique set of structure factor intensities after scaling and averaging symmetry-equivalent observations with
partialator, in three versions: fully merged as well as merged from the two complementary half-sets of equivalents - Text files
*.datwith crystallographic FOMs (S/N, CC_1/2, CC*, R_split) in resolution shells - A summary file written by the workflow tool, wrapping up indexing rates, unit cell refinement and FOMs
The summary file looks like this::
SUMMARY OF XWIZ WORKFLOW
Session time-stamp: 2020-07-22T12:18:48.711769
Operation mode:
interactive (run-time parameter confirm/override)
Input type:
virtual data set referring to EuXFEL-corrected HDF5
BASE CONFIGURATION USED
Group: data
path : /gpfs/exfel/exp/XMPL/201750/p700000/proc/
runs : 29
n_frames : 630000
... <ECHO OF CONFIGURATION FILE> ...
Step # d_lim source N(crystals) N(frames) Indexing rate [%%]
1 3.5 indexamajig 28553 630000 4.53
cell_check 28553 630000 4.53
2 2.0 indexamajig 27581 28553 96.60
OVERALL 27581 630000 4.38
Crystal unit cells used:
File Symmetry/axis, a, b, c, alpha, beta, gamma
hewl.cell tetragonal P c 79.1 79.1 37.9 90.00 90.00 90.00
hewl.cell_refined tetragonal P c 79.7 79.7 38.0 90.00 90.00 90.00
Crystallographic FOMs:
overall outer shell
Completeness 100.00 100.0
Signal-over-noise 5.84 2.54
CC_1/2 0.6338 0.1394
CC* 0.8363 0.4948
R_split 33.68 72.59